Sinh Học Hệ Thống
Ứng dụng học sâu trong sinh học đơn tế bào
- Chi tiết bài viết
- Bài viết liên quan
Giải trình tự từng tế bào (single cell RNA sequencing – scRNAseq) đã đưa khoa học sự sống của vài năm trước đây lên một tầm cao mới chưa từng có trong nghiên cứu sự khác biệt trong quần thể tế bào. Với tầm ảnh hưởng này, kỹ thuật giải trình tự tế bào đã được tập san khoa học Science tôn vinh là “thành tựu đột phá” (breakthrough) của năm 2018. Một trong những bước tiến lớn đó là sự nhận thức được rằng mặc dù các tế bào sinh học có vẻ giống nhau về mặt hình thái khi nhìn qua kính hiển vi, chúng có thể rất khác nhau về hệ gen biểu hiện, dẫn đến sự phân hóa về mặt chức năng giữa các tế bào.
Để nắm bắt được sự đa dạng của tế bào, cộng đồng khoa học Human Cell Atlas đề ra một mục tiêu đầy tham vọng. Đó là xây dựng một bản đồ toàn diện về hàng nghìn tỷ tế bào trong cơ thể người. Với sự phát triển của nền tảng kĩ thuật (ví dụ 10X Genomics), việc phân tích hệ RNA thông tin (transcriptome) của vài trăm tới vài triệu tế bào đã trở thành một lịch trình thường quy. Vì vậy scRNAseq (cùng với xử lý hình ảnh và genomics) trở thành một “kho” dữ liệu khổng lồ – dữ liệu lớn (Big Data) – một nguồn lực thống kê siêu dồi dào, mở ra một chân trời mới cho ứng dụng học máy (machine learning) và học sâu (deep learning) trong phân tích dữ liệu đơn bào.
Trong bài viết trên trang TowardDataScience, nhà tin sinh học Nicolay Oskolkov đưa ra cái nhìn tổng quan của mình trong lĩnh vực này, những thách thức trong mô tả phân tích toán học, và chỉ ra cách sử dụng học sâu với Keras và TensorFlow vào những vấn đề học không giám sát (unsupervised learning) trong phân tích dữ liệu RNA từng tế bào.
Tại sao sinh học đơn bào là nơi lí tưởng để áp dụng kĩ thuật học sâu?
Tiến hành một phân tích thống kê trên một số dữ liệu chúng ta phải hiểu sự cân bằng giữa
- số lượng đặc trưng p (gene, protein, biến thể di truyền, hay pixel hình ảnh)
- số lượng quan sát n (số mẫu, số tế bào, hay chuỗi trình tự).
Bộ gene của con người xấp xỉ p = 20 nghìn gene mã hoá protein trong khi gần đây công bố bộ dữ liệu 10X scRNAseq có tới n~1.3 triệu và n~2 triệu tế bào. Điều này ngụ ý rằng n >> p trong scRNAseq, đây là giới hạn điển hình trong học sâu. Khi làm việc trong giới hạn này, chúng ta phải vượt qua phân tính đại số tuyến tính và nắm bắt được cấu trúc phi tuyến tính trong dữ liệu. Đối với các giới hạn khác, khi n << p hoặc n ~ p, các phương thức như thống kê Bayes và Frequentist phù hợp hơn tương ứng cho từng loại. Nói cách khác, làm việc với dữ liệu scRNAseq chúng ta đối mặt với một vấn đề lớn, đó là: thông thường việc biểu đồ khớp quá mức (overfitting) và thiếu tính tổng quát là những mối bận tâm phổ biến trong sinh học tính toán. Đối với scRNAseq, chúng ta phải để ý tới sự không khớp đủ (underfitting), ví dụ như làm cách nào để sử dụng hầu hết dữ liệu.
Không chỉ scRNAseq bùng nổ gần đây trong khoa học sự sống mà những kĩ thuật đưa thêm nhiều loại thông tin khác của từng tế bào (OMICs trong thuật ngữ tin sinh học) cũng trở nên ngày càng phổ biến. Ví dụ như những tiến bộ gần đây trong nghiên cứu chromatin assessibility region (scATACseq), tạm dịch là vùng có thể truy cập được của sợi nhiễm sắc, mang lại bộ dữ liệu từ hơn 100K tế bào đơn. Trong khi một mình scATACseq không bảo đảm sự khám phá ra những quần thể tế bào hiếm (mục tiêu chính của phân tích dữ liệu đơn bào), nhưng nó cung cấp tiềm năng khổng lồ khi tích hợp với bộ dữ liệu thu từ scRNAseq do đó sẽ cải thiện độ chính xác trong việc chỉ định các tế bào thuộc vào một quần thể cụ thể nào đó.
Cuối cùng, các kỹ thuật đa-OMIC đơn bào (CITE-seq, scNMTseq và vân vân), đó là nguồn hội tụ đa thông tin từ cùng tế bào sinh học, dù vẫn chưa chạm tới kích thước mẫu đủ lớn điển hình cho scRNAseq nhưng cực kỳ hứa hẹn cho những thách thức tích hợp dữ liệu trong tương lai với học sâu.
Giảm chiều biểu diễn dữ liệu với học sâu
Bởi vì mục tiêu chính của phân tích scRNAseq là khám phá các quần thể tế bào mới, đây chính là một kiểu học không giám sát trong thuật ngữ học máy. Do đó, hai kỹ thuật phân tích quan trọng nhất được sử dụng cho scRNAseq là giảm chiều (kích thước) biểu diễn (dimensionality reduction) và phân cụm (clustering).
Bộ mã hóa tự động (Autoencoder) là một mạng neuron thần kinh nhân tạo thuộc nhánh học không giám sát với kiến trúc hình bướm để giảm kích thước. Trái ngược với kỹ thuật tuyến tính như phân tích thành phần chính (principle component analysis PCA), chia tỉ lệ đa chiều (multi-dimentional scaling MDS), hay phân tích nhân tố (factor analysis FA), bộ mã hóa tự động (Autoencoder) tự động thực hiện giảm kích thước phi tuyến tính và do đó có thể thu được cấu trúc phi tuyến tính cao của dữ liệu đơn bào.
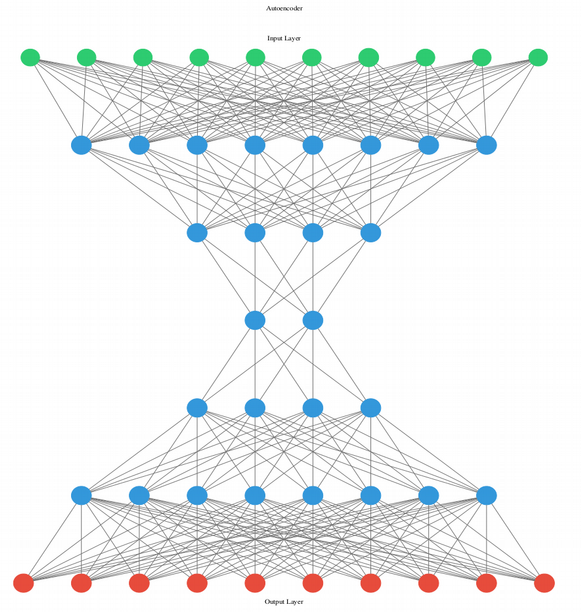
Trong ví dụ dưới đây, tác giả chứng minh sự khác biệt về độ phân giải đơn bào giữa kỹ thuật giảm chiều tuyến tính (PCA) và phi tuyến tính (Autoencoder) – sử dụng bộ dữ liệu CITEseq scRNAseq từ khoảng 8 nghìn tế bào máu cuống rốn đơn nhân (cord blood mononuclear cells) làm ví dụ. Tệp đầu vào ở đây là dạng bảng với các cột hàng dọc là các gene và cột hàng ngang là các tế bào, cột hàng dọc cuối cùng của tệp là chú thích “loại tế bào” thu được thông qua kỹ thuật phân cụm scRNAseq sử dụng. Tác giả gợi ý nên sử dụng kỹ thuật phân cụm biểu đồ với Louvaine community detection, cho phép xử lý dữ liệu đa chiều nhanh, phổ biến trong quy trình phân tích từng tế bào Seurat.
Để so sánh, tác giả cũng thêm biểu đồ t-distributed Stochastic Neighbor Embedding (tSNE) – đây hiện là kỹ thuật tiêu chuẩn trong giảm chiều trong lĩnh vực scRNAseq. Một vấn đề với tSNE là nó không thể xử lỹ dữ liệu nhiều chiều của scRNAseq. Do đó, phương pháp phổ biến là thực hiện PCA để giảm chiều trước sau đó kết quả tiền xử lý được đưa vào tSNE. Tuy nhiên, điều đó thậm chí có thể đạt được tốt hơn bằng cách thực hiện bước tiền giảm chiều theo cách phi tuyến tính với Autoencoder. Dưới đây là biểu đồ tSNE cho cả 2 chiến lược như vừa đề cập:
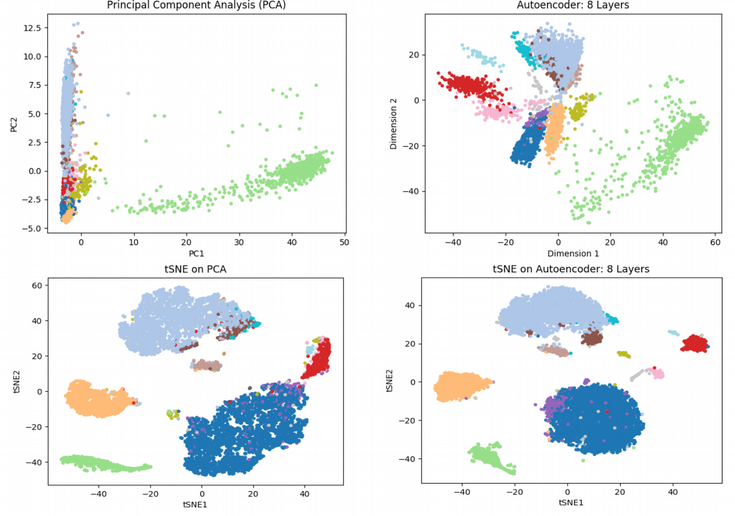
Trong biểu đồ này, mỗi điểm là một tế bào, màu sắc khác nhau tương ứng với loại tế bào khác nhau. Trong biểu đồ PCA, cho rằng bạn đang quan sát 12 quần thể tế bào, tuy nhiên bạn cơ bản không nhìn thấy chúng (tế bào chồng lấp dày đặc lên nhau) bởi vì giảm chiều tuyến tính không thể giải quyết cấu trúc tế bào đơn. Biểu đồ vẽ với Autoencoder trông tốt hơn, những quần thể tế bào khác nhau có thể phát hiện rõ. tSNE nhìn chung cung cấp các đốm tế bào sắc nét hơn. Tuy nhiên, trong trường hợp này, tSNE vẽ trên Autoencoder dường như có được các cụm dày và rõ hơn đặc biệt đối với cụm tế bào màu tím. Ở biểu đồ tSNE vẽ trên PCA, cụm màu tím bị phân tán với cụm xanh dương. Do đó, học sâu hứa hẹn sẽ cải thiện độ phân giải trong việc phát hiện các quần thể tế bào mới.
Tìm kiếm kĩ thuật giảm kích thước có khả năng mở rộng
Ngoài việc khó xử lý dữ liệu nhiều chiều, tSNE hoạt động kém khi số lượng tế bào lên tới hàng trăm nghìn và hàng triệu. FltSNE là phiên bản mới đầy hứa hẹn của Barnes-Hut tSNE, có khả năng xử lý tốt hơn với lượng dữ liệu khá lớn. Tuy nhiên, khi chạy FltSNE cho 1.3 triệu tế bào não chuột, tác giả thấy khó khăn khi lắp nó vào bộ nhớ. Đặc biệt trong trường hợp vẽ biểu đồ tSNE cho độ hỗn độn thông tin (perplexity) để kiểm tra cấu trúc toàn cục của dữ liệu. Tuy nhiên, độ hỗn độn thông tin perplexity = 350 là mức hỗn tạp tối đa có thể đạt được với cách sử dụng node với RAM 256GB trên cụm máy tính.
Phương pháp xấp xỉ và dự báo đồng nhất (UMAP) là một kỹ thuật giảm chiều phi tuyến tính khác có vẻ tốt hơn tSNE về nhiều mặt. Nó nhanh hơn tSNE, nhanh không kém FltSNE nhưng không cần nhiều bộ nhớ và dường như thu được cả cấu trúc dữ liệu scRNAseq cục bộ và toàn cục. Nhược điểm duy nhất mà tác giả thấy là có một chút không rõ ràng trong thuật toán đằng sau UMAP.
Gần đây, một vài phương pháp thú vị dựa trên bộ biến đổi tự động (Autoencoder) đã được đề xuất. Một trong số đó là SCVIS, một mạng lưới thần kinh (neural network) nắm bắt và hiển thị các cấu trúc ít chiều trong dữ liệu biểu hiện gen một tế bào, ngoài ra còn bảo tồn cả cấu trúc lân cận cục bộ và toàn cục.
Bên dưới tác giả đưa ra sự so sánh về bốn kỹ thuật giảm chiều, gồm PCA, tSNE/FItSNE, UMAP, và SCVIS dùng bộ dữ liệu 1.3 triệu tế bào chuột từ 10X Genomics:
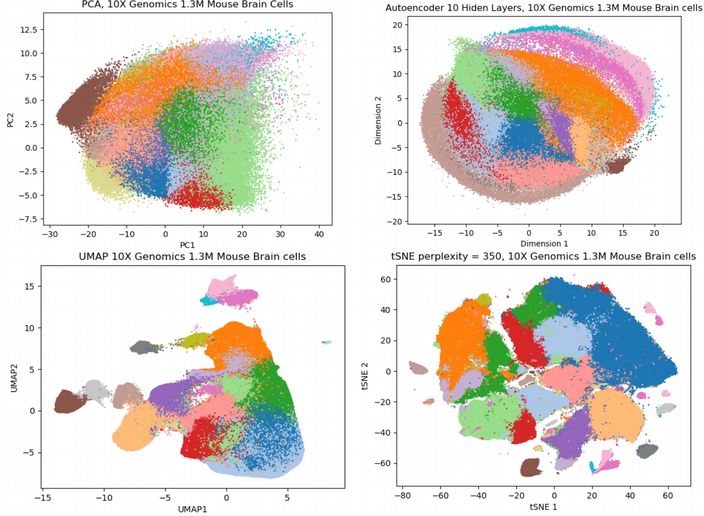
Đối với trường hợp CITEseq trên, ở đây chúng ta có thể thấy khác với PCA, ba kỹ thuật giảm chiều phi tuyến tính (SCVIS, UMAP và tSNE/FltSNE) đều có khả năng phân tách mọi quần thể tế bào có trong bộ dữ liệu scRNAseq. Trong ba kỹ thuật này, UMAP là cái nhanh nhất và thể hiện dữ liệu đã được giảm chiều khá tốt. So sánh về mặt thời gian tính toán, SCVIS mất khoảng 6 tiếng, FltSNE mất 3 tiếng và dùng rất nhiều bộ nhớ, và UMAP mất khoảng 3 tiếng khi chạy trên máy tính của tác giả. Nhìn vào lượng dữ liệu scRNAseq ngày càng tăng, tác giả dự đoán UMAP và Autoencoder sẽ thay thế tSNE trong tương lai.
Bộ tự động mã hóa sâu (deep Autoencoder) sử dụng dữ liệu scRNAseq với nền tảng Keras và TensorFlow
Trong hai phần cuối, tác giả hướng dẫn cách thực hiện từ đầu và chạy bộ tự động mã hóa sâu (deep Autoencoder) sử dụng Keras và TensorFlow. Bạn đọc có thể tìm thấy thông tin cụ thể trong về dòng lệnh (code) ở link này.
Bộ tự động mã hóa sâu (deep Autoencoder) sử dụng dữ liệu scRNAseq với nền tảng Keras
Như bạn sẽ thấy, nó không quá khó. Để tránh khó khăn khi đưa toàn bộ tập dữ liệu vào bộ nhớ, tác giả đã chọn 19 thành phần chính trên cùng mà tác giả thấy có ý nghĩa dựa trên việc tái khảo sát bộ dữ liệu, tức là xáo trộn các thành phần trong ma trận biểu hiện gene và kiểm tra tỷ lệ phần trăm phương sai được diễn giải bằng ma trận hoán vị (giả thiết không – null hypothesis). Bộ tự động mã hóa (Autoencoder) giảm dần chiều từ 19 xuống 2 (nút cổ chai của Autoencoder), mỗi lớp ẩn trong mạng thần kinh nhân tạo (neuron network) đang giảm xuống 1 chiều.
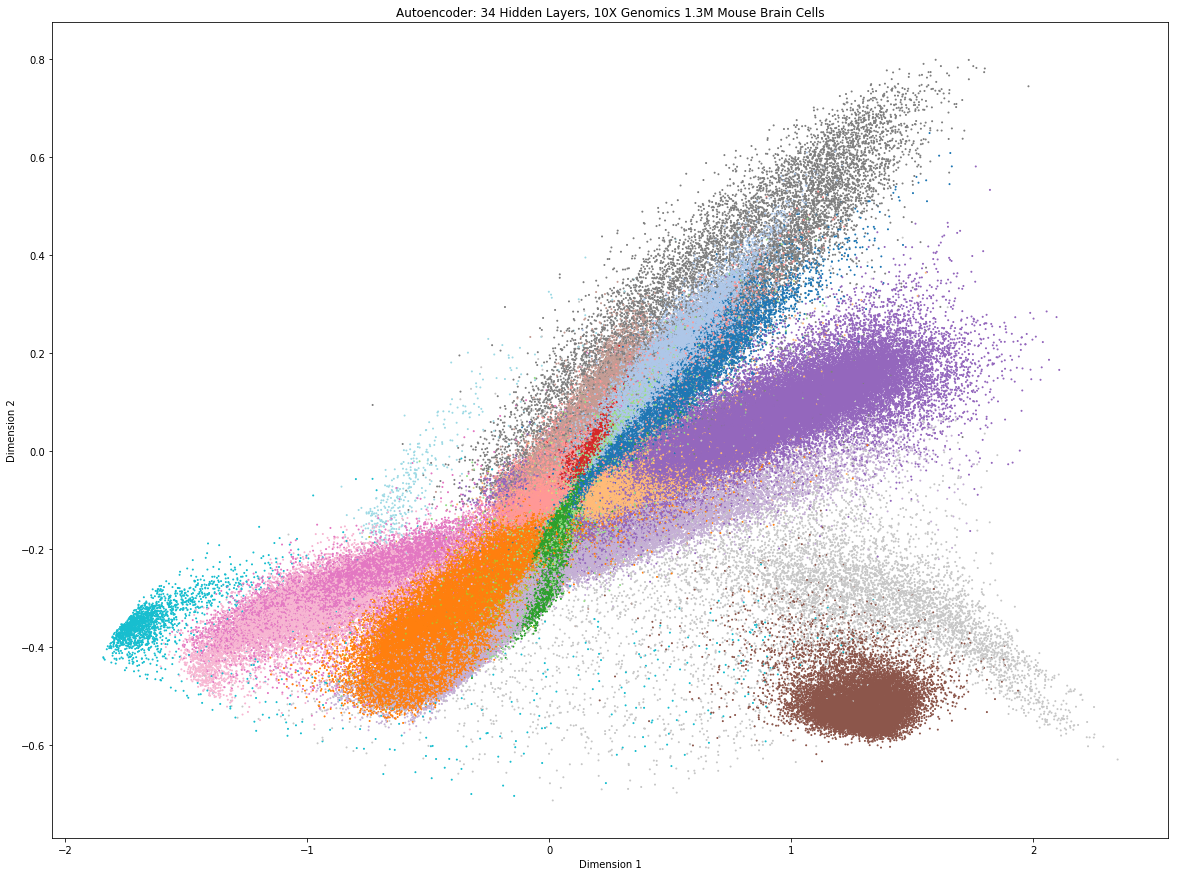
Chúng ta có thể thấy rằng những quần thể tế bào khá dễ phân biệt mặc dù tác giả chưa cố gắng tìm ra một cấu hình tối ưu của mạng neuron để cung cấp độ phân giải tốt hơn. Một lợi thế lớn của bộ tự động mã hóa sâu (deep Autoencoder) là thực thi rất nhanh và do đó có thể áp dụng với lượng lớn dữ liệu scRNAseq, chỉ mất khoảng hai phút chạy trên máy tính của tác giả cho mô hình để hội tụ và thu được biểu đồ trên.
Bộ mã hóa tự động sâu cho scRNAseq trên nền tảng TensorFlow
Keras sử dụng tiện lợi, hiệu quả, và nhanh, nhưng tác giả có ấn tượng rằng triển khai mạng neuron trên nền tảng TensorFlow cho người dùng sự kiểm soát tốt hơn. Ví dụ, người sử dụng Keras không bao giờ cảm nhận được cách kết nối thủ công giữa các nút neuron với Keras, trong khi điều này lại đạt được khi sử dụng với TensorFlow. Hệ quả là là thủ thuật hữu ích như học theo từng mẻ/tập nhỏ dữ liệu phải được lập trình bằng tay trong TensorFlow trong khi ở Keras lại có sẵn. Dưới đây là biểu đồ giảm chiều:
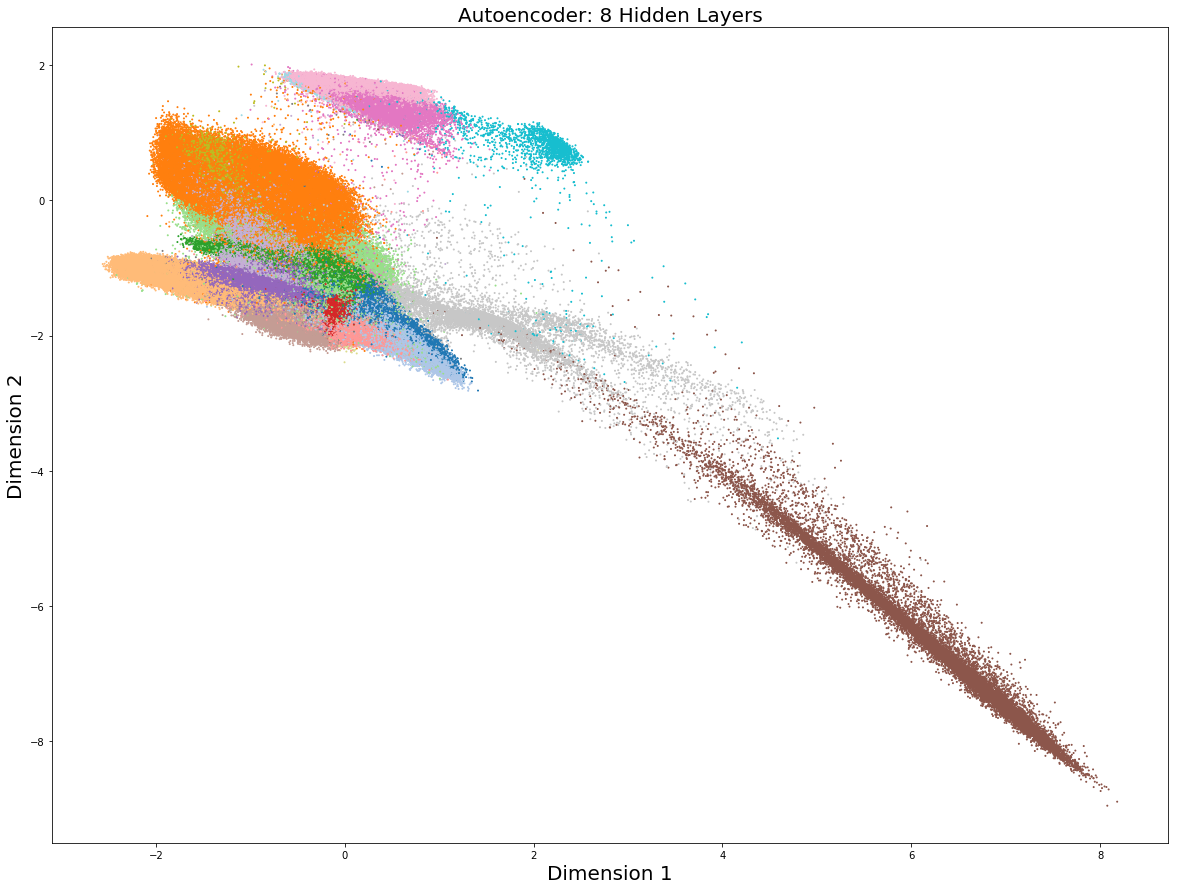
Một lần nữa, biểu diễn dữ liệu theo chiều thấp là có tiềm năng. Với một số điều chỉnh trong cấu hình và những tham số khác, người dùng sẽ có được độ phân giải tốt hơn rất nhiều. Tương tự như trong Keras, bộ mã hóa tự động (Autoencoder) dựa trên nền tảng TensorFlow hoạt động nhanh chóng, chỉ mất khoảng vài phút so với vài giờ khi sử dụng FItSNE, UMAP và SCVIS để vẽ biểu đồ có chiều đã được giảm. Nếu lịch trình tái khảo sát bộ dữ liệu là cần thiết vì nhiều lý do trong phân tích của bạn, nó sẽ không khả thi khi dùng với FItSNE, UMAP, và SCVIS nhưng sẽ rất dễ dàng khi tiến hành học sâu.
Tổng kết
Trong bài viết này chúng ta thấy rằng giải trình tự RNA đơn bào đã cải thiện nhanh chóng sự hiểu biết của chúng ta về sự đa dạng chức năng trong các tế bào sinh học. Giảm chiều (dimensionality reduction) có lẽ là công cụ phân tích quan trọng đằng sau mỗi quy trình phân tích scRNAseq. “Tiêu chuẩn vàng” của kỹ thuật giảm chiều tSNE hiện đang trải qua khó khăn trong khả năng mở rộng quy mô do lượng lớn dữ liệu được cung cấp từ công nghệ scRNAseq. Học sâu thông qua bộ tự động mã hóa (Autoencoder) và UMAP cung cấp những phương thức linh động và có khả năng mở rộng nhất hiện này để đạt được sự biểu diễn dữ liệu scRNAseq với chiều thấp và sẽ gần như có thể thay thế tSNE trong tương lai. Cuối cùng, chúng ta đã biết làm thế nào để thực hiện bộ tự động sâu cho bộ dữ liệu khổng lồ thông tin đơn bào của 1.3 triệu tế bào não chuột thu từ 10X Genomics cho Keras và TensorFlow.
Chuyển ngữ:
Nguyễn Hồng Nhung – Nghiên cứu sinh, Đại Học Myong Ji, Hàn Quốc
Nguyễn Tiến Tùng – Giảng viên, Đại Học Công Nghiệp Thành Phố Hồ Chí Minh, Việt Nam
Biên tập viên:
Dr. Trần Thị Hải Yến
Nguồn:
Nikolay Oskolkov. Deep Learning for Single Cell Biology. TorwardDataScience. 5 May 2019.