Công Nghệ Sinh Học
Đi tìm câu chuyện của từng tế bào: khó hay dễ?
- Chi tiết bài viết
- Bài viết liên quan
Sự sống của mỗi con người khởi nguồn từ một tế bào – hợp tử – mang bộ gen di truyền kết hợp của bố và mẹ. Tế bào đó trải qua quá trình phân chia, biệt hoá và chết tự nhiên để tạo nên hàng tỉ tỉ tế bào xây dựng nên một cơ thể hoàn chỉnh. Các tế bào được phân chia thành nhiều loại khác nhau tuỳ thuộc vào chức năng, hình dạng và nguồn gốc phát triển của từng loại. Nhóm các tế bào cùng đảm nhiệm một chức năng nhất định gọi là mô (tissue). Mặc dù cùng có chung một bộ gene, nhưng việc gene nào được biểu hiện và mức độ biểu hiện ít nhiều ra sao lại là câu chuyện của từng tế bào. Khi các nhà khoa học muốn có thông tin sinh học phân tử của tế bào đó, giải mã RNA (RNA-seq) là điều đầu tiên họ nhắm tới. RNA thông tin (mRNA) được phiên mã từ mã di truyền DNA và dùng để dịch mã ra trình tự amino acid cấu thành nên protein. Vì vậy, nếu biết được trong tế bào có các trình tự RNA thông tin nào, số lượng bao nhiêu là có thể biết một cách gián tiếp trong tế bào đó gen nào đang hoạt động tích cực, gen nào hoạt động cầm chừng, hay gen nào không hoạt động; cũng như dự đoán được sự có mặt của các loại protein. Tuy nhiên, để có một cái nhìn chi tiết và chính xác về hoạt động của tế bào, các nhà khoa học sẽ phải thực hiện thêm các thí nghiệm đánh giá trực tiếp hệ protein (proteomics), hệ mã DNA methyl hoá và histon code (epigenome) của tế bào đó.
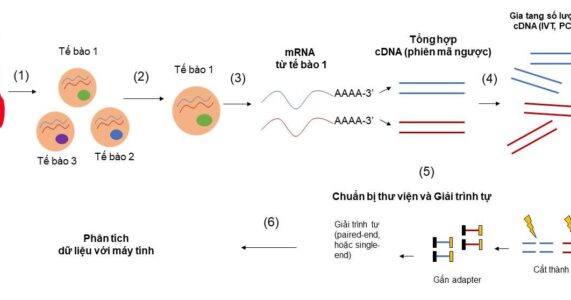
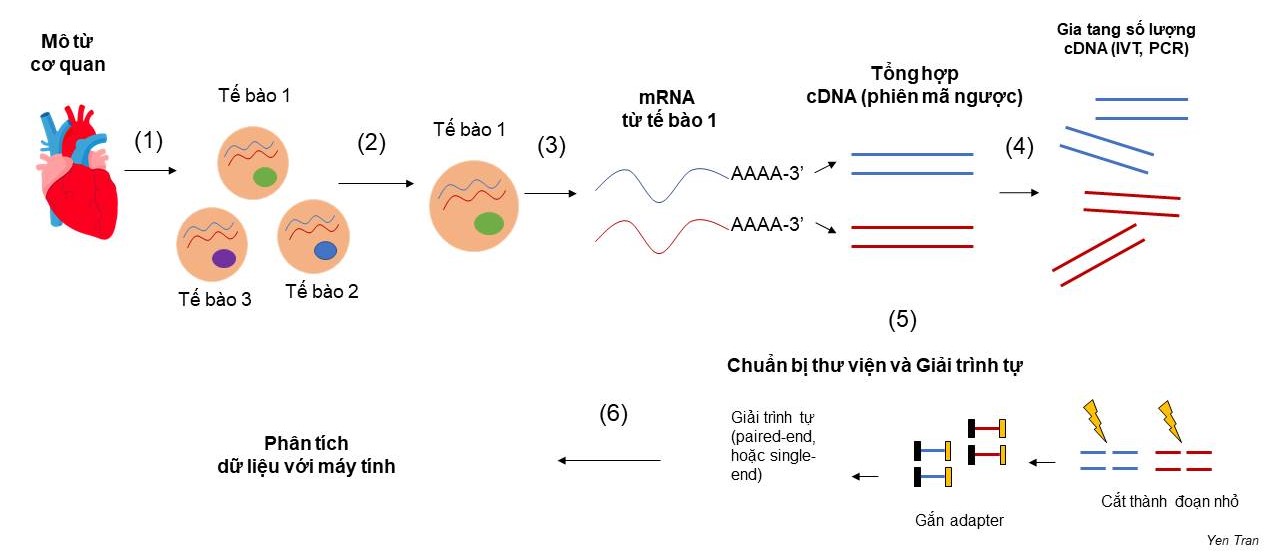
Với công nghệ kĩ thuật hiện tại, giải trình tự mRNA cho một tập hợp các tế bào (bulk RNA sequencing) không khó, thí nghiệm này đã rất phổ biến trong nhiều phòng thí nghiệm lớn nhỏ trên thế giới. Tuy nhiên, để giải trình tự mRNA cho một tế bào riêng lẻ, đó là bài toán không hề đơn giản, đòi hỏi nhiều phát minh đột phá, thúc đẩy các kĩ thuật sinh học phân tử lên tầm cao mới. Quy trình chung của “giải trình tự RNA thông tin từng tế bào” bao gồm (1) phân tách tế bào từ mô cơ quan, (2) phân tách từng tế bào từ một hỗn hợp tế bào, (3) tách chiết RNA của từng tế bào và tổng hợp cDNA (complement DNA), (4) Gia tăng số lượng cDNA (5) Chuẩn bị thư viện và giải trình tự (6) tính toán và phân tích kết quả (Hình 1). Trên phương diện kĩ thuật, bài viết này tập trung phân tích hai thách thức lớn nhất đặt ra ở bước 2 và 3:
Thách thức 1: Nếu lấy một mẫu mô nhỏ tới kích thước 1mmx1mmx1mm, thì cũng có ít nhất hơn mười nghìn tế báo cấu thành nên mẫu mô đó (tất nhiên số lượng tế bào còn phụ thuộc vào mẫu mô). Làm thế nào để phân tách từng tế bào riêng lẻ từ một nhóm tế bào để giải trình tự mRNA?
Thách thức 2: Càng nhiều tế bào riêng lẻ được giải trình tự, các nhà khoa học sẽ có càng nhiều dữ liệu để đánh giá về đặc điểm và tính chất của mẫu nghiên cứu. Làm thế nào để cùng một lúc có thể giải mã trình tự mRNA của nhiều tế bào riêng lẻ, cũng như làm thế nào để có đủ số lượng mRNA cho giải trình tự?
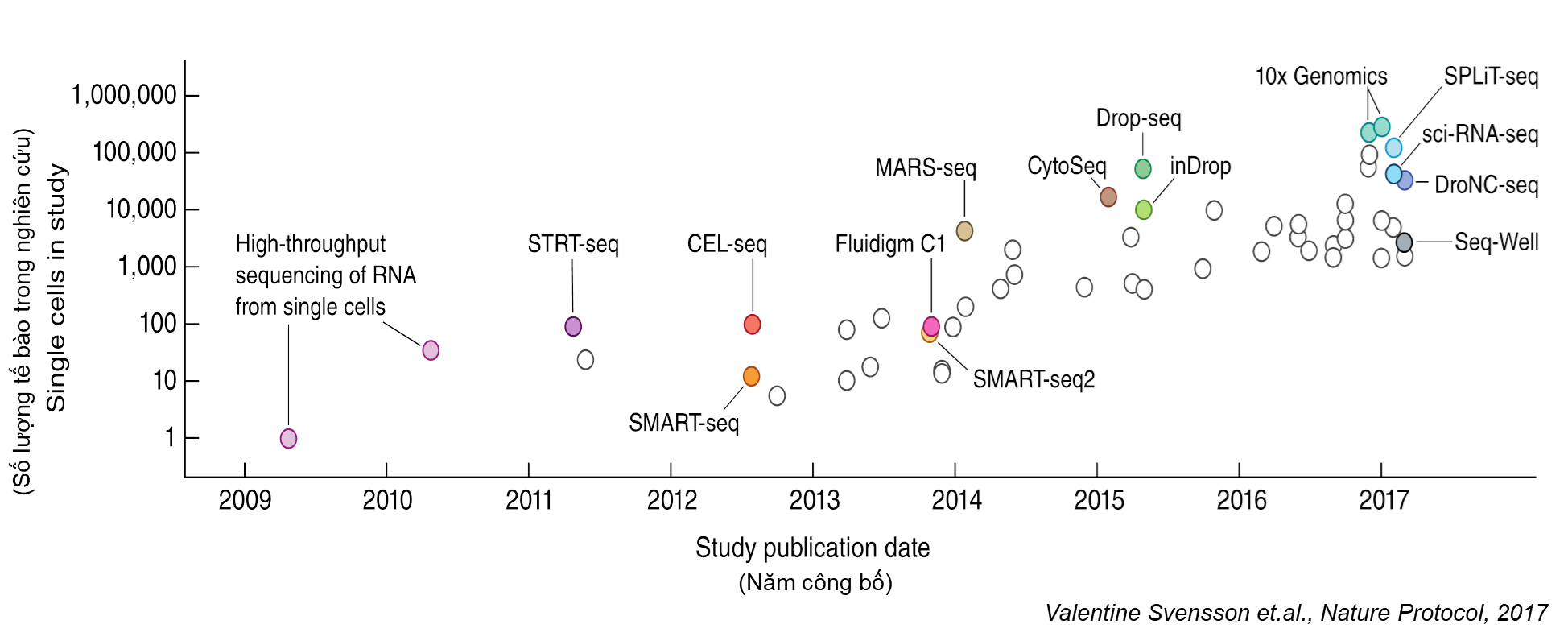
Từ năm 2009 cho đến nay, các phương pháp kĩ thuật công nghệ không ngừng được thay đổi, cải tiến và làm mới để giải quyết những thách thức của vấn đề nghiên cứu từng tế bào; tăng quy mô, giảm giá thành, đưa nghiên cứu từng tế bào trở thành một công cụ hữu ích giúp cho các nhà khoa học trả lời nhiều vấn đề hóc búa trong sinh học (Hình 2). Trong số các phương pháp đó, hai phương pháp đã được thương mại hoá và sử dụng rộng rãi nhất cho nghiên cứu từng tế bào là Fuildigm C1 và 10X Genomics.
Fuildigm C1 ứng dụng hệ thống vi thuỷ động học để dẫn dắt từng tế bào vào một buồng phản ứng riêng. Cốt lõi của Fluidigm C1 là một mạch chip gồm 96 buồng phản ứng với kích thước chỉ vừa đủ cho một tế bào, cùng hệ ống dẫn siêu nhỏ đưa tế bào đến buồng phản ứng và đưa sản phẩm ra khỏi buồng phản ứng (Hình 3). Khi dòng tế bào đi qua các buồng phản ứng này, lần lượt từng tế bào sẽ bị giữ lại. Ở mỗi buồng phản ứng sẽ có đầy đủ hoá chất cho các phản ứng tách chiết RNA và tổng hợp cDNA. Có hai nhược điểm lớn nhất của Fuildgim C1. Một là quy mô nhỏ, trong cùng một thí nghiệm, chỉ có tối đa 96 tế bào được phân lập và đánh giá. Hai là giá thành cao, nếu muốn phân tích 960 tế bào cùng một lúc, thì cần tới ít nhất là mười chiếc đĩa với giá thành khoảng 500-1000$/chip.

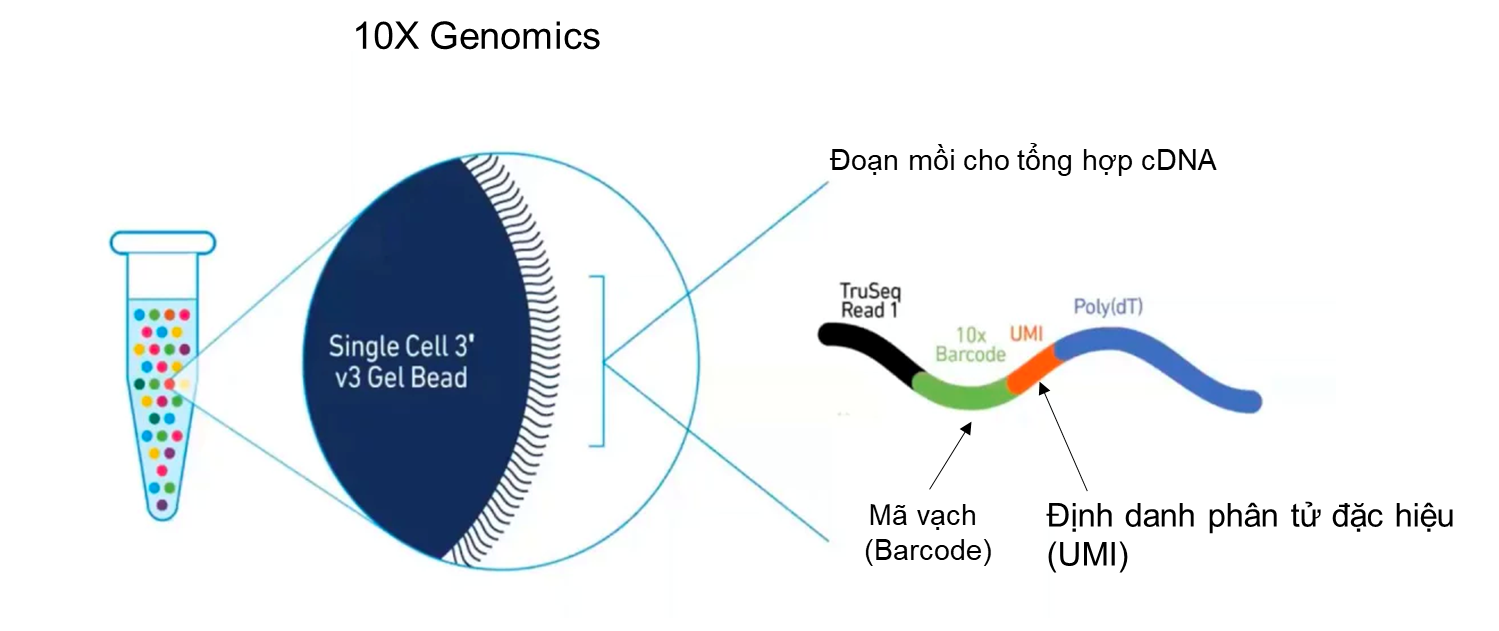
Sản phẩm của 10X Genomics ưu việt hơn Fluidigm C1 ở hai điểm này. Nguyên lý phân tách từng tế bào của 10X Genomics dựa vào hàm phân bố Poisson để điều khiển tốc độ dòng chảy tế bào và dòng chảy hạt hydrogel (hydrogel bead) gắn mã vạch (barcode) trên bề mặt, sao cho chỉ một tế bào và một hạt hydrogel được đóng gói trong một giọt dầu được gọi là GEM (Gel bead in Elmusion) (Hình 4). Với phương pháp này, một lúc có thể phân tích hơn 10,000 tế bào với giá thành giao động trong khoảng 5000$-7000$ cho một phản ứng đóng gói. Tuy nhiên, nhược điểm của kĩ thuật này là có thể có trường hợp một giọt dầu chứa hai tế bào hoặc hai hạt hydrogel. Các thông số về kĩ thuật, về số lượng nguyên liệu cho phản ứng phải được tính toán một cách cẩn thận để giảm thiểu tỉ lệ này xuống mức tối đa.

Để đánh dấu hệ RNA thông tin của từng tế bào, các nhà khoa học sử dụng phương pháp gắn mã vạch DNA vào cDNA của từng tế bào. Việc này có thể được thực hiện bằng cách chèn trình tự mã vạch vào đoạn mồi dùng để tổng hợp cDNA. Trong quy trình của Fluidigm C1, phản ứng phiên mã ngược từ RNA thông tin thành cDNA diễn ra ở từng buồng phản ứng riêng lẻ trong đó đã có chứa sẵn đoạn mồi mang mã vạch, đảm bảo mỗi tế bào có một mã riêng. Còn đối với 10X Genomics, đoạn mồi được gắn trực tiếp lên hạt hydrogel, ngay trong quá trình hình thành GEM, mỗi tế bào sẽ được đánh dấu nhận biết. Ưu điểm trong kĩ thuật đánh dấu của 10X Genomics là các GEMs có thể được gộp lại trước phản ứng tổng hợp cDNA, giúp thuận tiện trong các thao tác tiếp theo.
Để giải trình tự RNA, số lượng RNA tinh sạch phải đủ lớn, trong khoảng từ 100-1000 nanogram. Tuy nhiên, lượng RNA tinh sạch từ một tế bào lại quá nhỏ chỉ từ 1- 50 picogram tuỳ thuộc vào loại tế bào. Thế nên, trước khi giải trình tự, cần phải nhân bản thư viện mRNA hoặc thư viện cDNA tổng hợp từ mRNA lên đến một số lượng đủ lớn. Thư viện mRNA có thể tăng theo hàm tuyến tính bằng phương pháp “phiên mã trong ống nghiệm” (in-vitro transcription, IVT). Thư viện cDNA có thể nhân theo luỹ thừa của 2 bằng số chu kì của phản ứng PCR (polymerase chain reaction). Một nhược điểm có thể gặp phải trong phản ứng PCR là sự nhân lên thiếu cân bằng giữa các đoạn cDNA. Ví dụ, sau khi giải trình tự cDNA và so sánh với trình tự gene, nhận thấy số lượng cDNA của gene a nhiều hơn của gene b. Kết quả này có thể không phải do gene a biểu hiện nhiều hơn gene b mà do đoạn cDNA của gene a được nhân lên hiệu quả hơn so với đoạn cDNA của gene b trong phản ứng PCR.
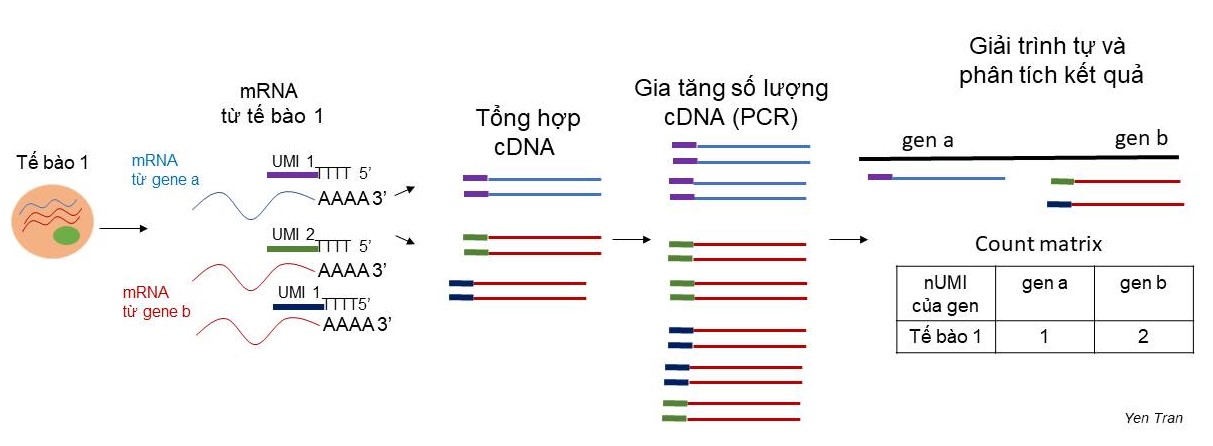
Để khắc phục nhược điểm này, trong đoạn mồi cho phản ứng tổng hợp cDNA, 10X Genomics có thêm vào một đoạn trình tự gọi là “định danh phân tử đặc hiệu” (Unique Molecular Identifier -UMI). Trình tự UMI được thiết kế sao cho mỗi UMI là đặc trưng cho một phân tử mRNA. Có khoảng 100,000 – 1,000,000 phân tử mRNA trong một tế bào ở động vật có vú. Trong đó có khoảng 10,000 phân tử mRNA khác nhau. Nghĩa là một gene có thể phiên mã tối đa khoảng 100 phân tử mRNA giống nhau. Theo tính toán, một đoạn UMI với 5 base pairs có thể đủ để phân biệt tới 1024 phân tử mRNA khác nhau (4^5= 1024). Trên bề mặt hạt hydrogel của 10X Genomics được gắn nhiều đoạn mồi có trình tự UMI khác nhau với số lượng đủ để phân biệt các phân tử mRNA (Hình 5). Sau khi cDNA được nhân lên, cắt nhỏ và giải trình tự, các đoạn cDNA có chứa UMI sẽ được so sánh với hệ genome và tính toán để biết số lượng UMI có trình tự khác nhau của từng gene là bao nhiêu, từ đó gián tiếp đánh giá mức độ biểu hiện của gene trong một tế bào (Hình 6).
Tính tới thời điểm hiện tại, kĩ thuật giải trình tự mRNA của từng tế bào đã đạt đến độ chín muồi. Có thể so sánh kĩ thuật này như một “chiếc kính hiển vi” có độ phóng đại lớn kết hợp với “chiếc máy ảnh” có độ phân giải cao giúp các nhà khoa học nhìn chi tiết hình ảnh của một tế bào ở mức độ phân tử, mở ra một lĩnh vực nghiên cứu mới – nghiên cứu sinh học từng tế bào.
Trần Thị Hải Yến (Biên soạn)
Nguyễn Tấn Trung (Hiệu đính)
Tài liệu tham khảo:
Bài báo:
Valentine Svensson et.al., Exponential scaling of single-cell RNA-seq in the past decade, Nature Protocol, 2017.
Atefeh Lafzi et. al., Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies, Nature Protocol, 2018.
Peter See et. al., A Single-Cell sequencing Guide for Immunologists, Frontiers in Immunology, 2018.
Saiful Islam et. al., Quantitative single-cell RNA-seq with unique molecular identifiers, Nature Methods, 2013.
Method of the Year 2019: Single cell multimodal omics, Nature Methods, 2020.
Eisentein, M. The secret life of cells. Nature Methods, 2020.
Website:
Video link:





